PBMs: Committed to Helping Patients
The Critical Path Forward: Rx
Policies to Reduce Patient Costs,
Improve Access
For America’s pharmacy benefit managers, PBMs, our core mission is to increase access to affordable prescription drugs for all Americans.
But more can be done and we stand ready to be part of the solution.
PCMA’s Policy Platform, outlined below, presents straightforward solutions that, taken together, would lower prescription drug costs and make pharmaceutical care even more accessible for all Americans.
This policy platform would result in total Federal Savings of $257.5 billion to $398.7 billion over ten years.
QUICK LINKS
PART ONE
PART TWO
PART THREE
PART ONE
Modernize Medicare Part D
In its first 15 years, Medicare Part D has significantly improved the affordability of prescription drugs for people with Medicare.
PBMs support a set of proposals, which, taken together, would modernize Part D and better align the incentives of all stakeholders to control drug costs and improve quality and outcomes for beneficiaries. These include policies to:
A) Improve Affordability
B) End Misaligned Incentives
C) Increase Choice and Competition
D) Keep Premiums Affordable
E) Achieve The Triple Aim
Modernize Medicare Part D
Total Federal Savings:
$280.3 billion over
ten years.
A) Improve Affordability
Unlike most patients with commercial drug coverage, Part D beneficiaries can face unexpected, high costs because
the Part D drug benefit does not have an annual limit on out-of-pocket spending.
We should cap annual out-of-pocket costs to provide better financial protection for beneficiaries with high drug costs
and make cost-sharing more affordable and manageable.
B) End Misaligned Incentives
Rather than lowering the increasingly high prices they set for their drugs, manufacturers want to shrink the discounts they owe Part D beneficiaries and taxpayers.
Instead, to lower drug prices and reduce costs, we should end misaligned incentives and hold manufacturers accountable for their high prices and require manufacturer contributions throughout the Part D benefit phase—from Initial Coverage to Catastrophic.*
*We believe that manufacturer responsibility should be higher in the Catastrophic Coverage phase and particularly for protected-class drugs and novel drugs without therapeutic competition and very high costs.

C) Increase Choice and Competition
The most important barrier to bringing costs down is the lack of competition and rules that stymie beneficiary choice and competition. By increasing competition, we can lower costs, increase access, and drive continued innovation in Part D.
We should build on Part D’s record of success by eliminating the two drugs per-class requirement; allowing select exclusions of the protected classes; encouraging beneficiaries to use lower cost drugs, such as generics and biosimilars; and promoting greater plan choices for beneficiaries.

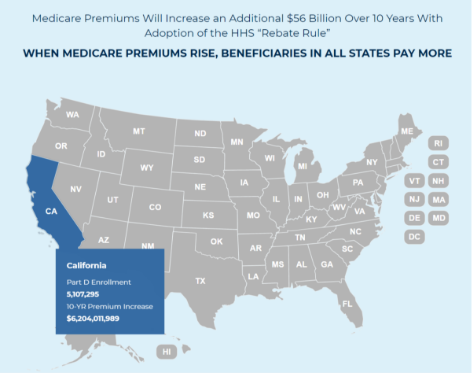
D) Keep Premiums Affordable
The rebate rule, which limits plans and PBMs from negotiating lower drug costs on behalf of Part D beneficiaries, would increase premiums 25% and cost taxpayers $170 billion.
A 2019 Government Accountability Office report found that PBMs passed 99.6% of all rebates on to plan sponsors, helping keep premiums and costs low. We should repeal the “rebate rule” and advance real solutions to bring down drug costs.
E) Achieve the Triple Aim
We should advance transformation in the delivery of pharmacy care services by rewarding value-driven pharmacies that promote quality care, improve clinical outcomes, and reduce out-of-pocket costs for Part D beneficiaries.
Rather than unravel value-driven gains, we can improve pharmacy pay-for-performance (“DIR”) by ensuring pharmacies know how they’re being measured, including through tools like EQuIPP, and requiring Pharmacy Administrative Service Organizations (PSAOs) to share with pharmacies the quality measures and criteria that they are required to meet.
PART TWO
End Anticompetitive Practices and
Enhance Competition
Stop Patent Abuse
Increased competition in the marketplace is essential for PBMs to effectively lower drug costs.
But brand drug manufacturer tactics that block generics and biosimilars from entering the market weaken PBMs’ ability to negotiate and deliver lower prescription drug costs. These pricing strategies, including “patent thickets,” “evergreening” and “product hopping,” unfairly protect some prescription drugs and biologics from more generic and biosimilar competition.
The result? Higher costs for patients and payers. We can do better by eliminating patent thickets, limiting evergreening and product hopping, and clearing the way for other manufacturers to bring their products to market, paving the way for more competition and lower costs.
- Eliminate anticompetitive “reverse payment settlements” or “pay-for-delay” agreements. Patent settlements, or “pay-for-delay” agreements, allow brand name drug and biologic patent holders to pay potential competitors to delay market entry or not produce a competing generic drug or biosimilar.
- Cap the overall number of years a biologic may enjoy cumulative patent protection.
- Target patent protection to truly innovative products.
- Codify the definitions of “product hopping,” “patent thicketing,” “evergreening,” and “secondary patent” to empower the Federal Trade Commission (FTC) to challenge these actions as anticompetitive.
- Apply stricter scrutiny to patent applications and thwart abuse by curbing “patent thickets.”
- Allow and provide additional funding for the FTC to use its equitable remedy authority to keep companies from engaging in patent abuse.
End Anticompetitive Practices and
Enhance Competition
Total Federal Savings:
Qbiosimilars
29 vs. 80
Currently there are only 29 Qbiosimilars on the market in the U.S. versus the 80 that are approved in Europe.
Reserve Market Exclusivities to True Innovation
Innovation in the prescription drug marketplace without affordability undermines patient access. Congress has granted over-long exclusivity periods for biologics and orphan indications leading to orphan-drug indication abuses and unforeseen delays in getting more affordable biosimilars to the market.
Reducing exclusivity periods for biologic medications and orphan drugs, along with reducing the first generic exclusivity period for certain non-first generics, will create more competition and lead to lower overall drug costs.
- End orphan drug exclusivity abuses. Orphan exclusivity periods should apply only to those prescription drugs originally approved by the U.S. Food & Drug Administration (FDA) under an orphan indication and only for the orphan indication itself.
- Revise innovator biologic exclusivity to seven years. Seven years (reduced from 12 years under current law) of market exclusivity would provide sufficient return for manufacturers while speeding competitor biosimilars to market to promote affordability and access.
- Examine whether FDA market and data exclusivities should be reformed, including reviewing how they are applied to ensure sufficient balance between rewarding scientific innovation and promoting competition and which types of products (like off-patent drugs) need additional exclusivity periods.
- Grant a five-year New Chemical Entity market exclusivity only if a product’s molecular structure contains a meaningful change from the existing drug.
- Require demonstration of greater clinical benefit for granting the New Clinical Investigation exclusivity.
- Limit the scope of the 180-day First-generic Exclusivity by allowing certain non-first generic applicants to obtain a court decision of patent invalidity or noninfringement to obtain immediate FDA approval.
Ensure Drugs Can Compete Fairly
Drug manufacturers, which alone set and raise prescription drug prices, use a host of strategies for fending off fair competition. These range from shadow pricing to abusing the Food & Drug Administration’s (FDA’s) citizen petition process to marketing schemes that undermine the tools employers and other health plan sponsors use to keep costs down for everyone.
While holding manufacturers accountable for those prices, policymakers could enforce antitrust laws, reform the FDA’s citizen petition process, and outlaw coupons (where they are not already considered a kickback) for drugs with direct competition. Eliminating tax deductibility for direct-to-consumer advertising of prescription drugs would also reduce inappropriate demand for higher-cost drugs.
- Hold pharmaceutical manufacturers accountable for their high drug prices.
- Enforce anti-trust laws to stop shadow pricing.
- Curtail “product hopping.”
- Foster competition by reforming the citizen petition process to curb their anticompetitive use.
- Advance real solutions to extremely high drug prices—not manufacturer marketing schemes, like coupons, which shift costs to other patients.
- Eliminate the tax-deductibility of direct-to-consumer prescription drug advertising.
Promote Generic and Biosimilar Competition
The key to reducing prescription drug costs is through increased competition in the marketplace. As more biosimilars enter the market, increasing their uptake will help boost competition and lower costs for patients. This can be achieved by streamlining how new biosimilars come to market, educating physicians and patients on the efficacy of biosimilars, making biosimilars interchangeable, and requiring a common billing code to make the substitution process smoother.
In addition, modifying cost-sharing for beneficiaries receiving low-income subsidies in Medicare Part D would encourage greater use of generics, lower cost brands, and biosimilars.
- Encourage growth of biosimilar market share via physician and patient education.
- Revisit the FDA’s nonproprietary label name guidance for interchangeable biosimilars to encourage switching or substitution.
- Require use of a common billing code to pay for a reference biologic and its biosimilars.
- Encourage the use of generics and other more cost-effective drugs in Part D. Allow CMS to modify cost-sharing for low-income subsidy beneficiaries to establish stronger incentives to select generics, lower cost brands, and biosimilars, with increasing levels of copayments for less cost-effective therapeutics options.
- Promote availability of biosimilars in Medicare Part D by ensuring Prescription Drug Plan sponsors have the flexibility to cover the biosimilar or the innovator (reference) product, but not require coverage of both, where there is a biosimilar in the class.
- Streamline the biosimilar development paradigm.
- Flip the burden of proving patent validity from the generic or biosimilar developer to the brand or biological maker.
- Enable biosimilars to launch without risk of treble damages.
- Allow for FDA accelerated approval of me-too brands. Accelerated review is granted by the FDA to new drug applications that address “unmet need,” the criterion for which should be updated to account for the economic need for competition to lower prices.
- Provide the FDA with sufficient resources to speed competition – particularly for lifesaving drugs and drugs with limited or no therapeutic competition (whether through generics or biosimilars).
- Require post-market surveillance and outcome reporting of patients who switch from a biologic to a biosimilar product.
PART THREE
Build a Value-driven, Equitable
Health Care System

Build a Value-Driven, Equitable Health Care System
Total Federal Savings:
PBMs are leading the effort to align reimbursement around the value of prescription drugs. Simply put, we should drive health care solutions that promote value and results, not volume.
PBMs and manufacturers negotiate value- and outcomes-based contracts for drugs that have quantifiable, widely-accepted outcomes. Data collected to inform these contracts continue to provide physicians and payers with insights that enhance clinical decision-making, improve patient health, and increase competition in the marketplace.
Accelerate Value-based Care
Greater adoption of value-based purchasing (VBP) and accelerating patient-focused pharmacy care can improve health outcomes. Providing Medicare Part D Prescription Drug Plans with access to Medicare Parts A and B claims data, establishing safe harbors for VBP contracting, and allowing Medicare Part D and state Medicaid plans greater flexibility to adopt private-sector formulary management techniques will provide a boost to these payment models.
Part D plans can utilize medical data in combination with prescription data to improve health outcomes. Legislative changes allowing the use of these data also would advance indication-based formularies and decrease prescriber burden.
Give Part D plans meaningful access to Part A and B claims data to coordinate care and make the best coverage decisions for beneficiaries, Part D plans need to be able to use medical data as well as prescription data. Legislative changes allowing use of these data for this purpose also would advance indication-based formularies and decrease prescriber burden.
Remove remaining barriers to the uptake of innovative payment and incentive structures that promote pharmaceutical value. While the Centers for Medicare & Medicaid Services (CMS) took a necessary first step with respect to the treatment of VBP arrangements under Medicaid Best Price, there are additional improvements that would foster greater innovation and expansion in the use of VBP for prescription drugs.
Regulatory amendments creating a safe harbor for VBP of prescription drugs would enable innovation in design and encourage participation, specifically:
- Create additional safe harbors for drug-related VBP at 342 C.F.R. § 1001.952;
- Expand the “value-based purpose” definition to include “reducing total cost of care of a target patient population” as a value-based purpose at 342 C.F.R. § 1001.952 and 42 § C.F.R. 411.351; and
- Expand the “value based entity participant” definition to include PBMs and, to the extent that such parties are participating in VBAs with PBMs and health plans, drug manufacturers, manufacturers of durable medical equipment, wholesalers, and distributors at 42 C.F.R. § 1001.952 and 42 §C.F.R. 411.351
Explore additional ideas through the Center for Medicare & Medicaid Innovation (CMMI) to expand on recent efforts to create and test new value-based payment models, including outcomes-based payment and Medicare Part B drug payment reform. Medicare Part D plans have been working with CMMI on innovative demonstration programs, including the new Part D Insulin Senior Savings Model. Other models similarly could be explored.
Expand the CMS Part D Payment Modernization Model to include additional value-driven flexibilities, such as facilitating patient use of high-quality, cost-effective medications by allowing select exclusions of certain classes from protected status.
Permit states the same tools available to employers and Medicare Part D plans to limit coverage of drugs for which there is inadequate evidence of clinical effectiveness or ample competition of therapeutic alternatives States should have the flexibility to adopt private-sector formulary management techniques, which drive value and lower costs, in their Medicaid programs.
Advance Use of Real-World Evidence
In a world where the cost of a given biologic can exceed an individual’s lifetime earnings, biopharmaceutical manufacturers should be expected to undertake ongoing research in their products, even after they are approved.
Health plans and PBMs need accurate, scientifically reliable information on prescription drugs throughout their lifecycles, from pre-approval for timely coverage decisions to post-market surveillance, and research into side effects and long-term efficacy for expedited approvals. These efforts should include rigorous evidence of drugs’ performance under real-world conditions.
Ensure Pre-approval Information Exchange (PIE) to ensure timely coverage of new drugs and indications. Codify current regulatory safe harbors that allow for PIE between manufacturers, plans, payers, and PBMs before FDA approval or authorization. PIE will also allow plan sponsors to better anticipate a new indication and plan for its impact on budget and patient care.
Authorize the FDA to assess value at the time of a drug’s approval or authorization (e.g., low additional value, high additional value, innovative and high value). If a drug is deemed by the FDA to be conditional or low additional value, that could be used by payers as a criterion for negotiating down net prices and/or negotiating outcomes-based contracting.
Accelerate efforts to build a robust real-world evidence (RWE) program and rigorous, science-based criteria for how RWE can be used to inform payment and coverage decisions (e.g., for use in VBA strategies).
Enforce and strengthen existing post-market surveillance requirements, particularly for expedited drug approvals involving small clinical trials, and require the FDA to share findings publicly to inform coverage decisions and value-based payment of prescription drugs.
Require manufacturers to continue research into the long-term efficacy and side effects of drugs under expedited approval (after they come to market) with specific timelines and reporting requirements.